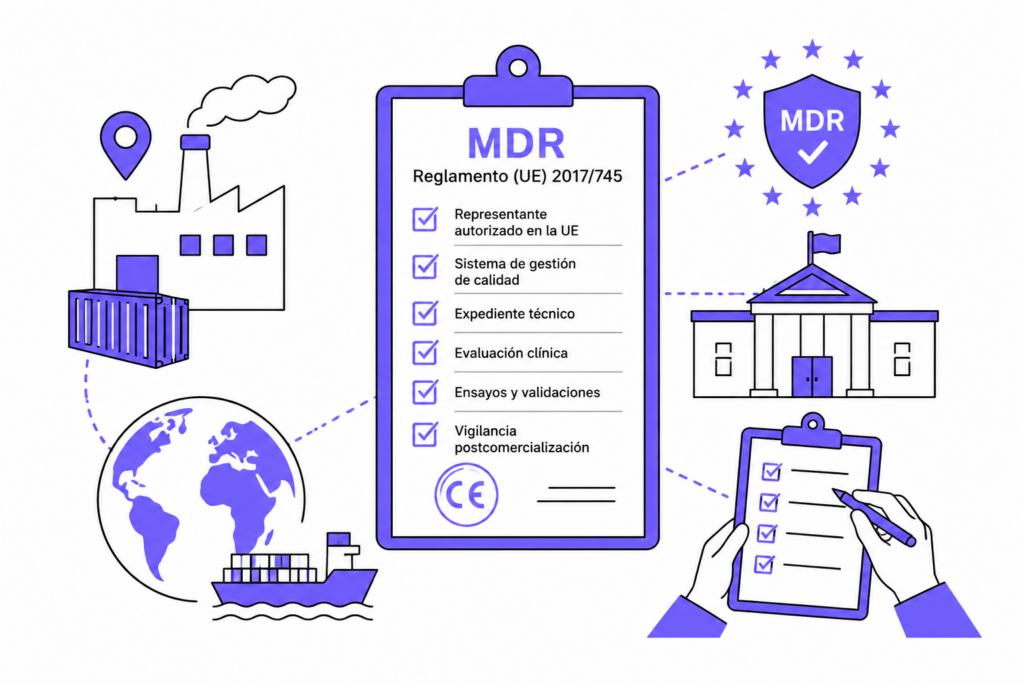
El Reglamento (UE) 2017/745, conocido como MDR (Medical Device Regulation), ha transformado por completo el panorama regulatorio de los dispositivos médicos en Europa. Este marco normativo sustituye a las antiguas directivas (MDD 93/42/EEC y AIMDD 90/385/EEC), imponiendo requisitos más estrictos para garantizar la seguridad, trazabilidad y transparencia en todo el ciclo de vida del producto sanitario.
Para los fabricantes, comprender y aplicar correctamente el MDR es esencial para mantener el acceso al mercado europeo. En este artículo, te explicamos de manera práctica qué implica este reglamento, cuáles son sus principales exigencias y cómo un laboratorio especializado como Med-Lab IBV puede ayudarte a cumplirlo con eficacia.
¿Qué es el Reglamento MDR 2017/745?
El MDR (Medical Device Regulation) es la normativa que regula desde el diseño y fabricación hasta la comercialización y seguimiento poscomercialización de los dispositivos médicos en la Unión Europea. Aunque se aprobó en 2017, no entró plenamente en vigor hasta 2021, tras sucesivos periodos de transición para permitir la adaptación de los fabricantes a las nuevas obligaciones.
A diferencia de las antiguas directivas, el MDR, al tratarse de un reglamento, es de obligado cumplimento en en todos los Estados miembros, sin necesidad de transposición nacional.
Su objetivo es claro: reforzar la seguridad del paciente y aumentar la confianza del mercado mediante una evaluación técnica y clínica más rigurosa.
Principales novedades del MDR
El MDR ha introducido un enfoque mucho más exigente en la validación de los dispositivos médicos. Entre los cambios más relevantes destacan:
🔹 1. Revisión de la clasificación de los dispositivos
Los productos se clasifican ahora en clases I, IIa, IIb y III, según su nivel de riesgo. Muchos dispositivos que antes se consideraban de bajo riesgo (como ciertos implantes o productos reutilizables) han sido reclasificados, lo que implica nuevos requisitos de ensayo y certificación.
🔹 2. Evaluación clínica reforzada
Se exige evidencia clínica sólida que demuestre la seguridad, eficacia y rendimiento del dispositivo. Los fabricantes deben realizar evaluaciones preclínicas y ensayos biomecánicos o funcionales, así como estudios postcomercialización para asegurar su comportamiento a largo plazo.
🔹 3. Trazabilidad y vigilancia postventa
El MDR introduce el sistema UDI (Unique Device Identification), que permite rastrear cada dispositivo desde su fabricación hasta su uso final. También obliga a los fabricantes a establecer un plan de vigilancia postcomercialización y un sistema de gestión de riesgos actualizado de forma continua.
🔹 4. Requisitos más estrictos para los organismos notificados
Los organismos notificados (Notified Bodies) encargados de otorgar el marcado CE deben cumplir criterios más rigurosos de independencia y competencia técnica, lo que aumenta la fiabilidad del proceso de certificación pero también puede prolongar los plazos de evaluación.
🔹 5. Transparencia y documentación técnica ampliada
Se exige una documentación técnica más detallada, con justificación científica de cada parámetro evaluado, y la publicación de información relevante en la base de datos EUDAMED, accesible para autoridades y usuarios profesionales.
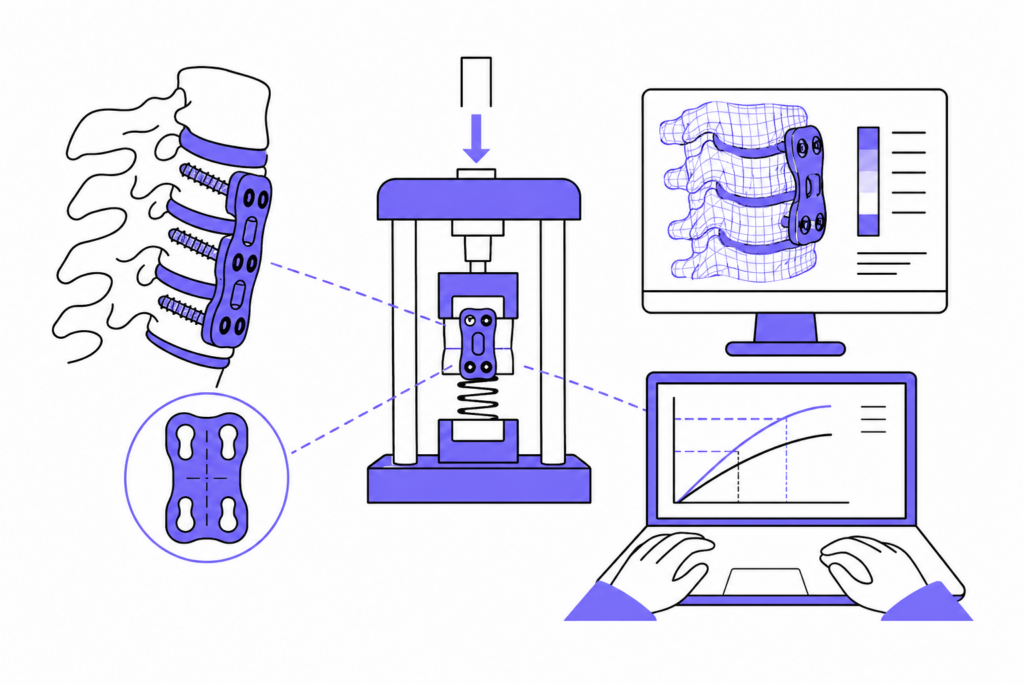
Ensayos técnicos y preclínicos exigidos por el MDR
El MDR establece que los dispositivos deben demostrar su conformidad mediante evidencia técnica validada. Esto incluye ensayos mecánicos, funcionales y de biocompatibilidad que garanticen la seguridad y el rendimiento del producto antes de su introducción en el mercado.
Algunos de los ensayos más habituales son:
- Ensayos de fatiga y resistencia estructural en implantes (ISO 14801, ISO 7206, ASTM F382…).
- Pruebas de flexión, torsión y compresión para placas, vástagos y prótesis.
- Evaluaciones biomecánicas en entornos simulados que reproducen las condiciones reales de uso.
- Análisis de biocompatibilidad y toxicología según la serie ISO 10993.
Conoce cómo realizamos estas pruebas en nuestro laboratorio acreditado de ensayos mecánicos para dispositivos médicos, donde aplicamos las normas ISO/IEC 17025 y MDR 2017/745 para garantizar resultados fiables y aceptados por organismos notificados.
Cómo afecta el MDR a los fabricantes
El impacto del MDR va más allá de los ensayos. Supone una transformación completa en la gestión del ciclo de vida del dispositivo.
🔸 Nuevas responsabilidades del fabricante
El MDR exige la designación de una Persona Responsable del Cumplimiento Regulatorio (PRRC) dentro de la empresa. Además, impone obligaciones en cuanto a vigilancia, trazabilidad, y reporte de incidentes adversos.
🔸 Mayor inversión en documentación técnica
Los expedientes deben incluir información sobre diseño, materiales, ensayos, gestión de riesgos, evaluación clínica y post-market surveillance. Un expediente incompleto puede implicar retrasos en la obtención del marcado CE o incluso la retirada del producto del mercado.
🔸 Revisión más rigurosa por parte de los Organismos Notificados
Los organismos notificados están aplicando criterios más estrictos y auditando con mayor profundidad los procesos de ensayo y validación de los fabricantes. Trabajar con un laboratorio acreditado bajo ISO/IEC 17025 simplifica esta auditoría, ya que sus resultados son reconocidos oficialmente.
El papel de los laboratorios acreditados en el cumplimiento del MDR
Los laboratorios especializados desempeñan un papel esencial en la demostración de conformidad. Un laboratorio acreditado actúa como tercero independiente que valida el cumplimiento técnico del producto con las normas aplicables.
En este sentido, Med-Lab IBV ofrece un valor diferencial:
- Acreditación ISO/IEC 17025 reconocida por ENAC.
- Amplia experiencia en ensayos de implantes quirúrgicos, exoesqueletos y ayudas técnicas.
- Metodologías alineadas con MDR, ISO 13485 y normas ASTM/UNE-EN.
- Interpretación técnica y asesoramiento regulatorio incluidos en cada informe.
De este modo, los resultados emitidos por Med-Lab IBV no solo cumplen con los requisitos normativos, sino que aceleran los procesos de evaluación y certificación CE.
Ejemplo práctico: de la prueba mecánica al cumplimiento regulatorio
Imaginemos un fabricante que desarrolla una prótesis de cadera de nueva generación. Según el MDR, deberá aportar evidencia de:
- Seguridad mecánica → Ensayo de fatiga y resistencia según ISO 7206-4.
- Durabilidad y estabilidad funcional → Ensayo de carga angular conforme a ASTM F384.
- Evaluación biomecánica del movimiento → Validación funcional en laboratorio especializado.
- Cumplimiento normativo → Documentación técnica respaldada por resultados trazables y acreditados.
Med-Lab IBV realiza todas estas pruebas bajo un mismo marco técnico y regulatorio, garantizando que el producto cumpla los requisitos de rendimiento y seguridad exigidos por el MDR.
Del cumplimiento a la competitividad
El MDR 2017/745 no debe verse como un obstáculo, sino como una oportunidad para reforzar la calidad, la confianza y la competitividad de los fabricantes europeos.
Cumplir con esta normativa demuestra compromiso con la seguridad del paciente, transparencia en la cadena de suministro y excelencia técnica en el diseño del producto.
En Med-Lab IBV, acompañamos a fabricantes, ingenieros y responsables de calidad en todo el proceso de validación técnica, ayudándoles a reducir plazos de certificación y asegurar el cumplimiento normativo con garantías.
Contacta con Med-Lab IBV para planificar tus ensayos conforme al MDR 2017/745 y optimizar tu estrategia de validación y acceso al mercado europeo.